Cost modelling comparison of adherent multi-trays with suspension and fixed-bed bioreactors for the manufacturing of gene therapy products
Cell & Gene Therapy Insights 2019; 5(11), 1663-1674.
10.18609/cgti.2019.175
There is huge pressure on biomanufacturing facilities due to high demand for gene therapy products such as recombinant adeno-associated viral (AAV) vectors made under current good manufacturing practice (GMP) conditions. The constraints of the scale-up process are rarely taken into consideration during clinical process development, with many facilities simply scaling-out adherent cell stack protocols, procedures, and test methods. In order to demonstrate the importance of cost economics linked to the choice of scale-up processes, this study provides a detailed cost modeling analysis comparing viral vector production in adherent cells in cell stacks to viral vectors produced in bioreactors, either in suspension using a stirred bioreactor or adherently in fixed-bed bioreactor. The results show that single-use bioreactors allow the cost of installed capital and labor to be reduced significantly and that fixed-bed bioreactors with optimized production protocols offer the greatest experimental robustness and the best opportunity for increased productivity in a manufacturing facility.
Introduction
In recent years, many gene therapy products have moved towards the clinic and today there are approximately 3000 products in clinical trials, of which approximately 122 are in Phase 3 [1], more than 200 involve adeno-associated viral (AAV) vectors. As recombinant AAV combine high transduction efficiency and long-term expression with safety and low immunogenicity, they are viral vectors of choice for in vivo gene therapy applications. As a result, their wider use in clinical trials has underscored the need for improved production protocols and manufacturing processes in order to yield sufficiently large amounts of highly pure recombinant AAV particles [2,3]. Transient transfection of plasmids remains the most widely used method for clinical manufacturing of AAV. It is usually performed in adherent HEK293 (human embryonic kidney 293 cells) or HEK293-T cells grown in adherent systems such as multi-trays (MT) (also called cell-stacks or cell factories depending on the supplier), or roller bottles using a transfection reagent such as CaPO4 or PEI. Although still widely used, this method is time-consuming and labor-intensive when scaling up; therefore several alternative production platforms are being developed: one of them is transfection in suspension [4]. For this protocol the HEK293 cells are adapted to grow in suspension in animal-free and antibiotic-free media. Transfection in suspension is performed in flasks, rocking platform or stirred-tank bioreactors, which are easier to handle compared to numerous multi-trays; however, one drawback is reduced transfection efficiency compared to adherent systems, which can lead to lower viral titers [2]. As a result, another option has emerged, combining both ease of handling with the ability to transfect adherent cells on a large scale, namely fixed-bed bioreactors such as the iCELLis® bioreactor. The iCELLis® 500+ bioreactor from Pall Biotech (Figure 1
Methodology
In this study, we have performed a comparative cost modelling analysis of three upstream production (USP) processes for AAV production namely transfection of adherent cells in multi-trays, transfection of cells in suspension using single-use bioreactor (Figure 2
This analysis presented here will facilitate decision making around which technology to use to generate virus for commercial production. The methods were compared in models generated using BioSolve Software (Biopharm services), a widely used software for costs analyses in the biopharmaceutical industry. This software is used not only for cost reduction and increased profitability, but also for improved planning and rapid and effective scale-up. It generates a comprehensive overview of a factory environment, including capital expenses (CAPEX) such as footprint, and capital investment and operating expenses (OPEX) including media, buffer and reagent usage (including plasmid DNA), process duration and workload, as well as consumables in USP. The analysis also took into account the downstream production (DSP) process and quality controls (QC), in order to give a realistic view of cost of goods (CoGs). The cost structure was determined, and different scenarios were compared using the software to identify parameters that can affect the CoGs. The model assumed 100% utilization of the manufacturing facility.
Results
Cost model assumption & existing data
The traditional adherent multi-tray-based process (MT) was compared to a cell suspension process performed in a single-use bioreactor (STR bioreactor) and to the suspension cells trapped in the fixed-bed process, the iCELLis® bioreactor (Pall Biotech). In order to allow a direct comparison between the bioreactors and avoid possible bias, we choose to model the bioreactor systems using the same suspension seed train. For all processes, both the clinical scale (200 L) and the manufacturing scales (800–1000 L) were evaluated.
In the iCELLis bioreactor, cell densities are correlated with metabolites profiles. This correlation is established at small scale during process development and validated at large scale.
In all cases, a cell lysis with the same lysis buffer was performed to release and harvest the vector. The Benzonase treatment was performed just after the cell lysis step by adding 0.05% v/v of Benzonase in the lysed harvest, at the end of the USP production step.
QC controls were averaged at $15000 per batch (Table 1). QC tests do not differ from one scenario to the other. The QC tests consider AAV quantification by Q-PCR and capsid ELISA for full/empty capsid ratio, bioburden, HCP, residual DNA, turbidity, pH, metabolites, etc. (partial list). For the sake of the comparison, the quality of the product was assumed to be similar across all scenarios.
| Table 1: Parameters used for throughput and cost modelling of three processes at clinical and manufacturing scales. | ||||
|---|---|---|---|---|
| MT process | Suspension bioreactor | iCELLis Bioreactor | Optimized iCELLis Bioreactor | |
| Seed train | MT10 | Shake flasks and single use (SU) stirred bioreactor | ||
| Production mode | MT10 ($500/MT10) | SU stirred bioreactor | iCELLis 500 bioreactor | iCELLis 500 bioreactor |
| Culture media | Seed:10% serum ($50/L); Prod: 2% serum ($30/L) | Serum-free media ($90/L) | Serum-free media ($90/L) | Serum-free media ($90/L) |
| Clinical volume | 200 L | 200 L | 200 L (133 m2) | 200 L (133 m2) |
| Manufactured volume | 1000 L (1 L/MT10) | 1000 L | 800 L (333 m2) | 800 L (333 m2) |
| Transfection cell density | 160,000 cells/cm2 | 1 x 106 cells/mL | 160,000 cells/cm2 | 200,000 cells/cm2 |
| pDNA/cell (μg/106 cells) | 1.5 ($100,000/g*) | 1.5 ($100,000/g*) | 1 ($100,000/g*) | 0.8 ($100,000/g*) |
| rAAV titer in USP | 2 x 1013 vg/L (2 x 104 vg/cell) | 2 x 1013 vg/L (2 x 104 vg/cell) | 2.5 x 1013 vg/L (2 x 104 vg/cell) | 3.125 x 1013 vg/L (2 x 104 vg/cell) |
| DSP | Same unit operations for all process – yield = 30% Affinity capture: binding capacity 5 x 1012 vg/mL (resin: $25,000/L) | |||
| Dose definition | Dose = 1 x 1014 vg | |||
| QC cost | 15,000 $/batch | |||
| Processes rely on single-use technologies for USP, media and buffer preparation. *Pricing based on high volume orders of GMP-grade plasmid. | ||||
Figure 3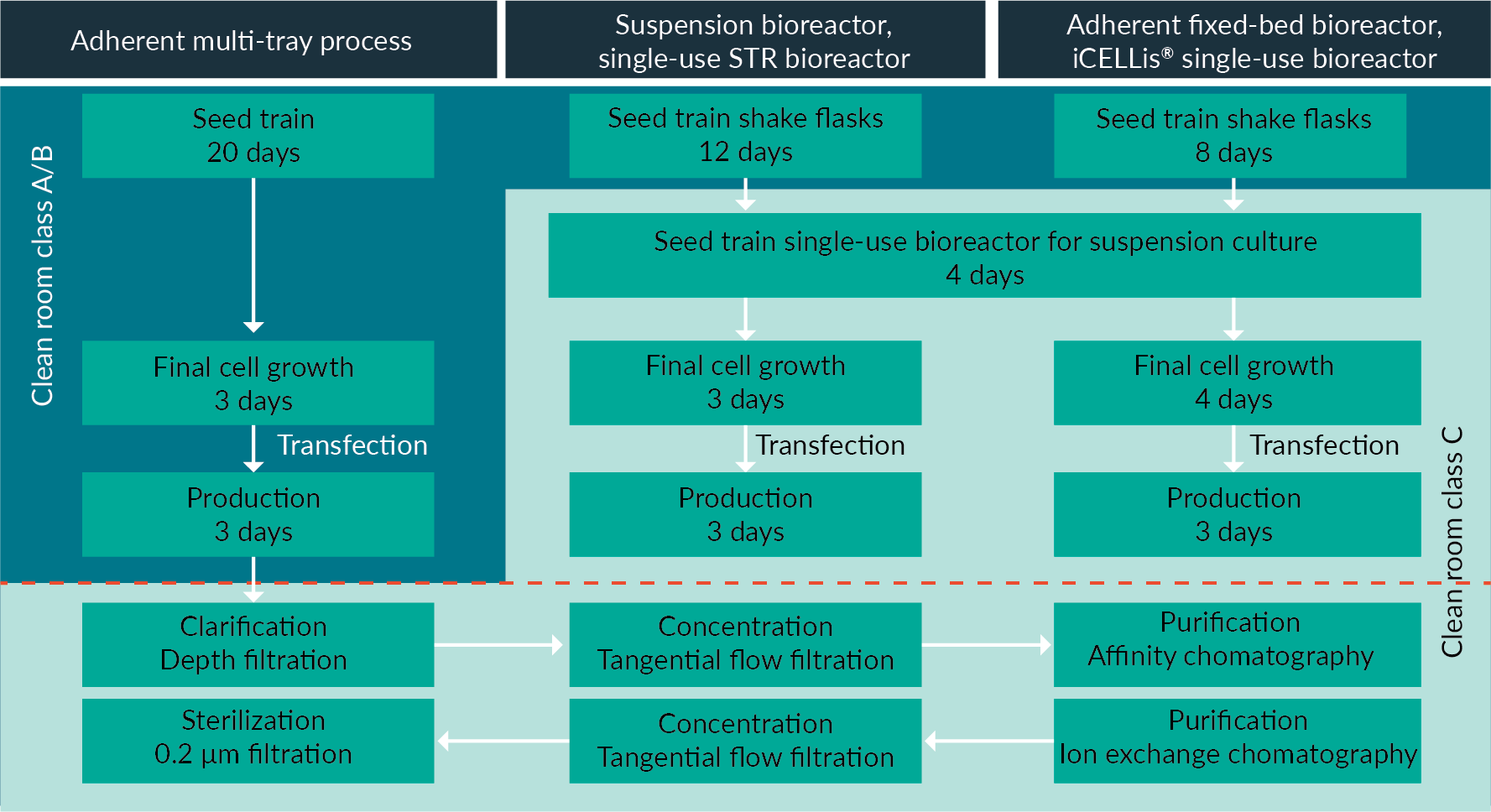
The DSP process, which is also included in the modeling, is shown in Figure 3. An overall 30% recovery yield was applied for the complete DSP process whatever the scenario, in order to better compare the different USP platforms. There are a few small differences in the operational procedures. For example, the clarification step for the suspension culture requires two filters, whilst the fixed-bed bioreactor only requires one. This is due to the fixed bed acting as a trap for cell debris thereby reducing the turbidity of the supernatant, so only one filtration step is needed for clarification. Whilst these small differences may have an operational impact, the overall impact on economic modelling of the DSP is negligible.
The parameters used for the throughput and cost modeling of the three processes at clinical and manufacturing scales are described in Table 1. The analysis integrated the cost of consumables and equipment. The clinical scale for all processes was 200 L; and the manufacturing scale was 1000 L for the cell stacks (corresponding to 10 MT10 with 1 L of cell culture medium each) and the suspension bioreactor. For the iCELLis® bioreactor, it was 800 L. As it is a fixed-bed bioreactor, the volume of media in the vessel only ranges from 60 to 80 L, with the remaining media circulating in an adjacent tank. The circulating volume of media can reach up to 1000 L, however in this system, the ratio of media volume to cell surface can easily be adjusted, often resulting in optimized protocols using lower amounts of media. Therefore, for the modeling analysis, we chose to use 800 L of circulating cell culture for the manufacturing scale in the iCELLis® bioreactor, as this protocol produces comparable viral titers to the suspension bioreactor.
The media/surface ratio for the MT scale was smaller than the iCELLis volume/surface ratio (1.5 L/m2 vs 2.4 L/m2), because the MT platform works with media exchanges, when in the iCELLis bioreactor these are usually avoided. The transfection process was PEI based with the same ratios applied across scales and scenarios. The quantity of DNA per million of cells for each scenario is detailed in Table 1.
The production efficiency in the iCELLis® bioreactor is comparable to that in a suspension bioreactor using lower cell density and consuming less media. Upon further optimization, a protocol with higher cell density at the time of transfection, requiring the addition of a lower concentration of DNA per cell, shows that the viral titer can be increased further. Optimization of the process in the iCELLis® bioreactor therefore has a significant impact on production yields and costs, suggesting that fixed-bed bioreactors can offer a robust and economical solution for viral vector manufacturing.
The USP AAV titers detailed in Table 1 were based on an average of customer’s having compared adherent and suspension processes and PALL’s experience. Higher titers in the iCELLis bioreactor have been obtained, but these were not applied for this work.
Some of the CoGs is also indicated in Table 1 to give an overview of the cost distribution. Of interest is the quantity of GMP-grade plasmid DNA (pDNA), an essential reagent for transient transfection, which makes up a large portion of the cost of GMP production at around $100,000 per g. This pricing is based on high volumes of GMP-grade reagent ordered. Media costs and consumables are also included.
Cost calculations & analysis
The number of doses produced per year and the CoGs calculated using the Biosolve software are shown in Figure 4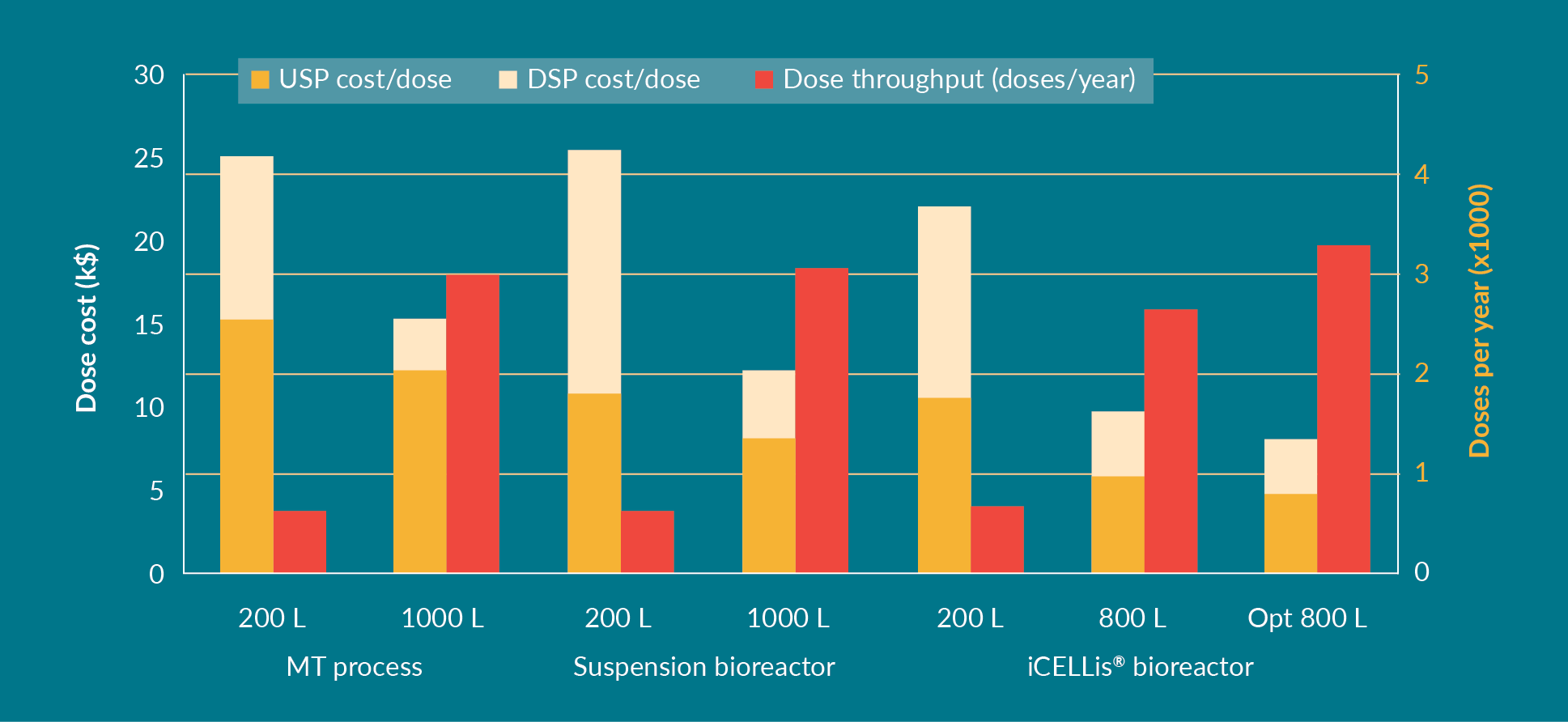
For the MT process at the 200 L scale, the cost per dose (USP and DSP combined) was $25 k, decreasing to $15 k during scale-up to 1000 L. For the suspension process, the cost per dose was comparable to MT in 200 L but became more cost-effective at the 1000 L scale ($12 k). For the iCELLis® bioreactor process, the initial cost per dose at clinical scale was lower compared to MT and suspension ($21 k), decreasing to $10 k per dose at the 800 L scale – and even less after protocol optimization ($8 k). Although the number of doses produced per year is less in the iCELLis® bioreactor than in the suspension bioreactor, due to the longer bioreactor process and the immobilization time in the bed after cell seeding, the iCELLis® bioreactor still offers the most cost-effective scaling up process, especially after optimization. With an optimized protocol, a higher amount of viral vector is produced using the same total amount of DNA, which in turn, results in a lower cost per dose. Therefore, with an increase in the number of doses per batch due to optimization, the overall number of doses per year is also higher.
Further expenditure analyses including CAPEX and OPEX for each process are shown in Figure 5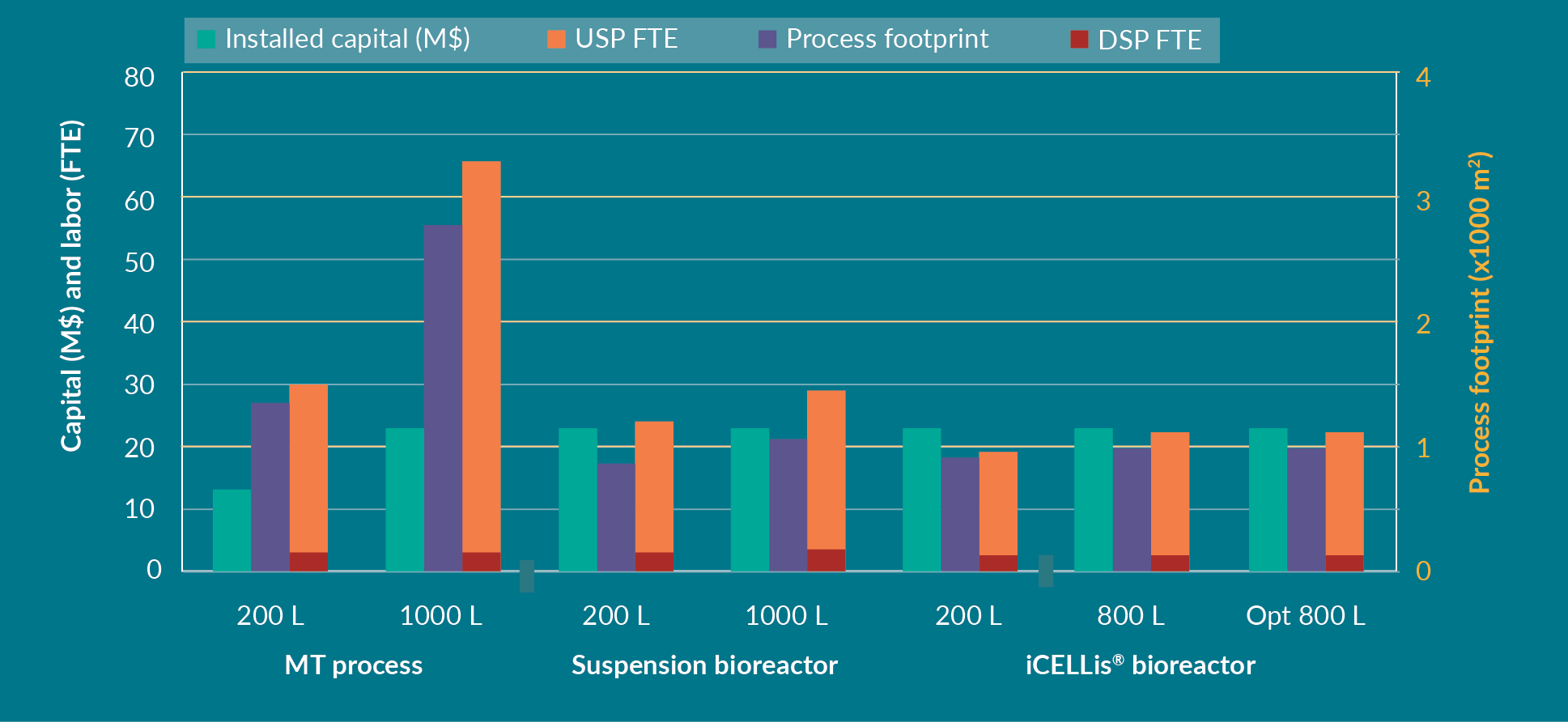
Benchmark process modeling
Benchmark process modeling is represented in Figure 6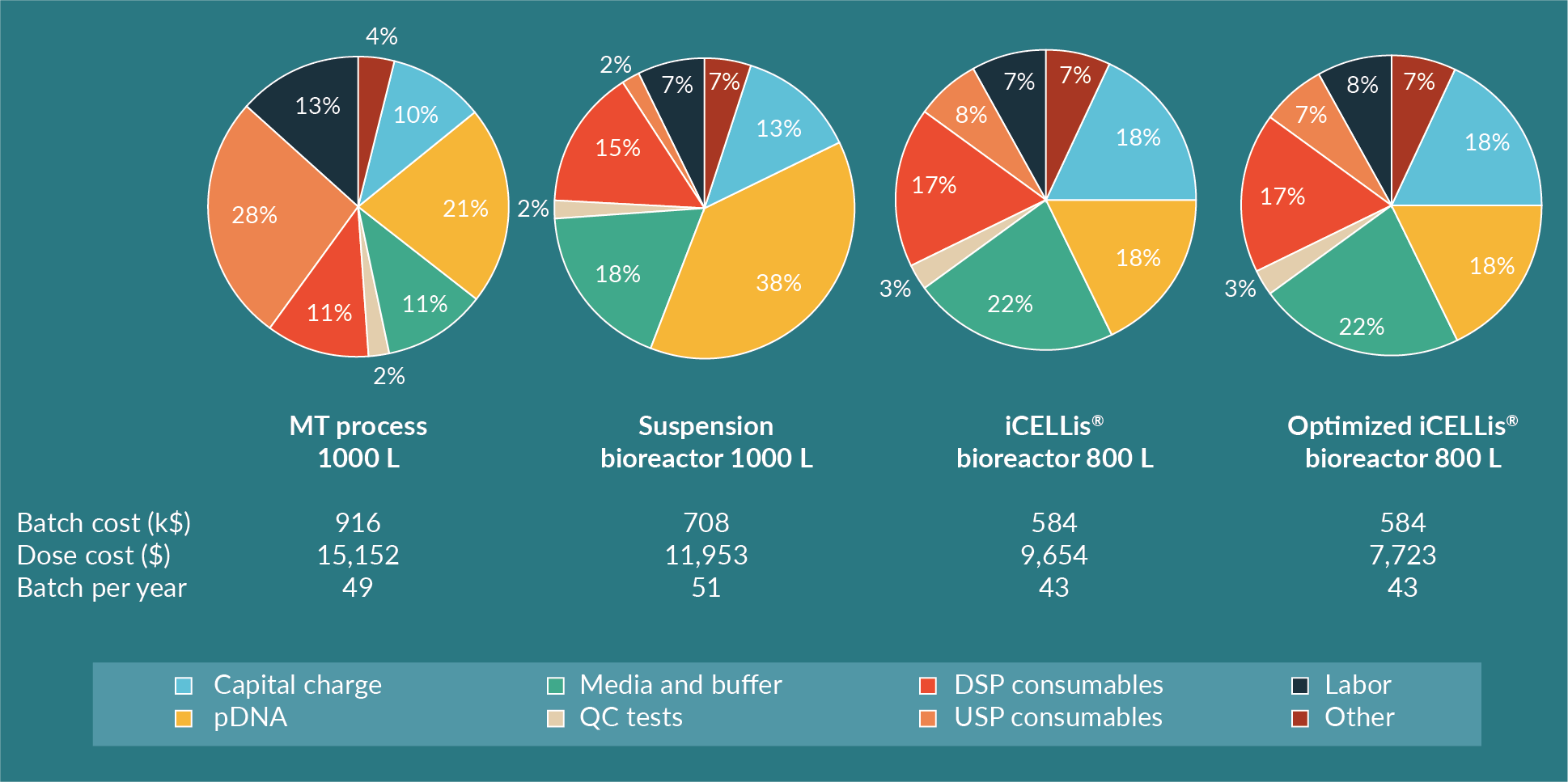
In the iCELLis® bioreactor, the CAPEX is higher compared to the suspension bioreactor, mostly because the hardware is more expensive, and the single-use vessel is also more costly than a single-use biocontainer needed in a stirred bioreactor. Nevertheless, the cost per dose and per batch is lower in the fixed-bed bioreactor in this modeling analysis. The limitation of the iCELLis® bioreactor remains the lower number of batches per year compared to the other processes due to the slightly longer production time (1 day longer than suspension bioreactor) for cell growth. Depending on the operational process of the facility and the overall manufacturing capabilities, a second iCELLis® bioreactor maybe required if more batches are needed. Another option is to increase the cell surface in the bioreactor itself for higher yields of AAV. This is likely to be the favored option as usually a set number of doses are needed rather than a set number of batches. In this case, the iCELLis® bioreactor allows the production of 3200 doses per year after protocol optimization, which is more than the MT and suspension processes.
De-risking the choice of USP technology
The cost per dose is a key element in the strategic choice between the different production processes and the comparison is shown in Figure 7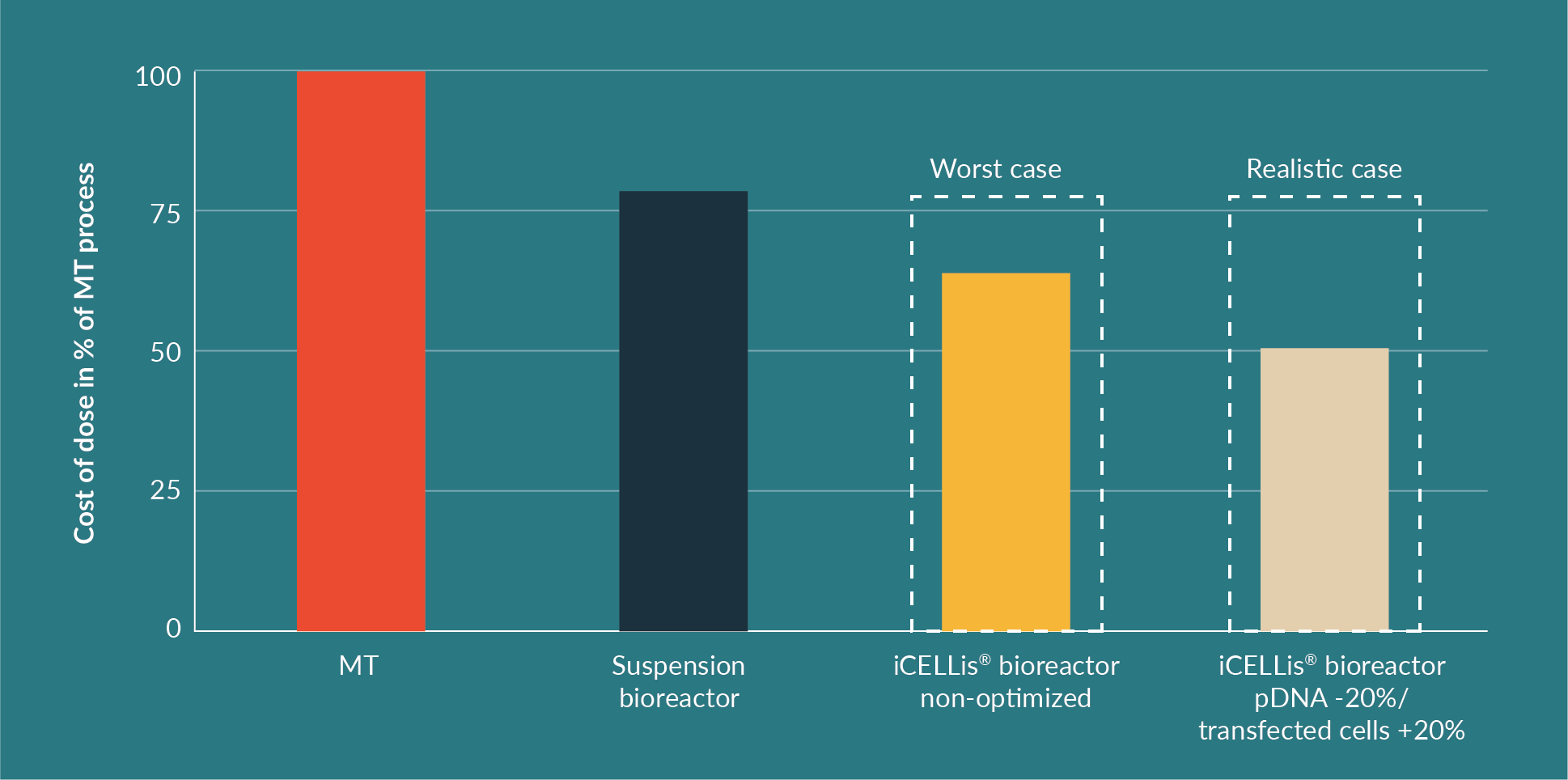
Whilst cost may seem to favor suspension bioreactors, handling and experimental set-up does not. Homogeneous transfection in 1000 L volume suspension bioreactors is challenging mostly due to handling issues (volumes and amounts of DNA used) and reproducibility issues, which are often encountered due to cell homogeneity affecting the transfection process. Thus, through experimental optimization, the iCELLis® bioreactor process increases productivity and is likely to outperform the suspension process for the clinical production of AAV using transient transfection.
Another significant factor in choosing which technology to use for upstream production of virus is the time needed for process development. For the treatment of some rare genetic diseases with a small number of patients, time to market is critical and process development can often be the rate limiting step. Adapting cells to an adherent bioreactor such as the iCELLis® system takes less than 3 months, and the process development from bench scale to industrial scale usually less than 1 year. In contrast, converting adherent processes to suspension can take up to a year for transient transfection processes. Developing packaging/producer cell lines for clinical production usually takes considerably longer (~2 years) without any guarantee of high titers. Thus, an adherent bioreactor represents a lower risk choice with lower cost for commercial manufacturing.
Conclusion & perspectives
Through a cost comparison and production cost analysis of three processes (adherent in multi-trays, suspension in a single-use bioreactor and adherent in a fixed-bed single-use bioreactor), this study emphasizes the cost-inefficiency of laboratory scale adherent multi-tray/cell stack systems. Thus, production in MT does not offer an affordable large-scale manufacturing process for AAV production for gene therapy. Although the process is still economically viable at the clinical scale, a development of processes with higher productivity and improved cost-effectiveness is recommended. Alternatives such as the use of high-producing suspension cells in single-use bioreactors allows significant cost reductions of installed capital and labor. However, the cost of GMP-grade plasmid DNA is still high, representing up to 40% of total batch cost and limiting the possibility of further reducing the CoGs. Overall for any process, transient transfection at 1000 L is challenging due to handling limitations and costly due to the quantity and cost of plasmid DNA required. As a result, the use of a fixed-bed bioreactor is an attractive option for further process optimization as it enables increased productivity by reducing the amount of DNA required for transfection. This approach is being used by several groups including the Saint Jude’s Hospital, who previously reported AAV production levels at a small scale in iCELLis® Nano bioreactor comparable to a MT control [6].
Further process optimization remains to be explored, including the use of an AAV packaging cell line as an alternative to transfection, and process operation in perfusion mode as described previously [7]. Applied at a large scale for AAV vector production, the latter would also take advantage of the iCELLis® bioreactor’s built-in perfusion capacities [8]. In the next years, the use of packaging cell lines for AAV manufacturing will increase, thus probably reducing the overall manufacturing costs by avoiding the current transfection step.
In summary, we and other groups have shown that the iCELLis® bioreactor platform not only offers experimental flexibility and robustness, but also a significant cost reduction opportunity for the production of adeno-associated viral vectors [5]. As a result, we expect a wider adoption of these systems in manufacturing facilities, where higher demand and increased environmental constraints require increased productivity and cost reduction. This will be supported by additional modeling studies for other retroviral vectors such as lentiviruses.

Authorship & Conflict of Interest
Contributions: All named authors take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Acknowledgements: None.
Disclosure and potential conflicts of interest: The authors declare that they have no conflicts of interest.
Funding declaration: The authors disclose that Pall Corporation owns patents relevant to the Biotech Industry.
Article & copyright information
Copyright: Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0 which allows anyone to copy, distribute, and transmit the article provided it is properly attributed in the manner specified below. No commercial use without permission.
Attribution: Copyright © 2019 Pall Corporation, Pall France SAS, Pall Belgium BVBA, and Pall Europe Limited. Published by Cell and Gene Therapy Insights under Creative Commons License Deed CC BY NC ND 4.0.
Article source: Invited; externally peer reviewed.
Submitted for peer review: Nov 20 2019; Revised manuscript received: Dec 12 2019; Publication date: Dec 18 2019.
References
1. Phases of Gene Therapy Clinical Trials; Journal of Gene Medicine: Website
2. Clement M, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016; 3: 16002. CrossRef
3. Robert MA, Chahal PS, Audy A, Kamen A, Gilbert R, Gaillet B. Manufacturing of recombinant adeno-associated viruses using mammalian expression platforms. Biotechnol. J. 2017; 12: 1600193. CrossRef
4. Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017; 8: 87–104. CrossRef
5. Pinhao Miessner. Production of AAV Vectors for Gene Therapy: a Cost-effectiveness and Risk Assessment Master thesis MIT, 2016.
6. Powers AD, Piras BA, Clark R, Lockey T, Meagher MM. Development and optimization of AAV hFIX particles by transient transfection in an iCELLis® fixed bed bioreactor. Hum. Gene Ther. Methods 2016; 27(3): 112–21. CrossRef
7. Grieger JC, Soltys SM, Samulski RJ. Production of Recombinant Adeno-associated Virus Vectors Using Suspension HEK293 Cells and Continuous Harvest of Vector From the Culture Media for GMP FIX and FLT1 Clinical Vector. Mol. Ther. 2016; 24(2): 287–97. CrossRef
8. Lesch HP, Heikkila KM, Lipponen EM et al. Process Development of Adenoviral Vector Production in Fixed Bed Bioreactor: From Bench to Commercial Scale. Human Gene Ther. 2015; 26(8): 560. CrossRef
Affiliations
Emmanuelle Cameau
Pall Biotech, Reugelstraat 2. B-3320 Hoegaarden, Belgium
Andreia Pedregal
Pall Biotech, Reugelstraat 2. B-3320 Hoegaarden, Belgium
Clive Glover
Pall Biotech, 5, Harbourgate Business Park, Southampton Road, Portsmouth PO6 4BQ,
United Kingdom
![]()